The Role of Herpes Simplex Virus 1 (HSV-1) in Alzheimer's Disease: A Pioneering Frontier for Therapeutic Innovation
Alzheimer's disease (AD), the most prevalent form of dementia, has long been linked to genetic and environmental factors. However, emerging research now positions Herpes Simplex Virus 1 (HSV-1)—a ubiquitous neurotropic virus infecting over 3.7 billion people globally—as a critical player in AD pathogenesis. HSV-1 establishes lifelong latency in trigeminal ganglia but reactivates under stress or immunosuppression, traveling to the brain via axonal transport. The virus directly destroys neurons through lytic replication, rupturing lysosomes to release proteases like cathepsins that degrade synaptic proteins and cellular structures. Indirectly, HSV-1 hijacks host machinery to promote endoplasmic reticulum (ER) stress and mitochondrial dysfunction, while viral glycoproteins (e.g., gB and gD) disrupt blood-brain barrier integrity. These mechanisms collectively trigger cascades of neuronal death, synaptic loss, and neuroinflammation—cornerstones of neurodegenerative diseases like AD. This revelation opens unprecedented opportunities for researchers to pioneer targeted therapies addressing this understudied mechanism.
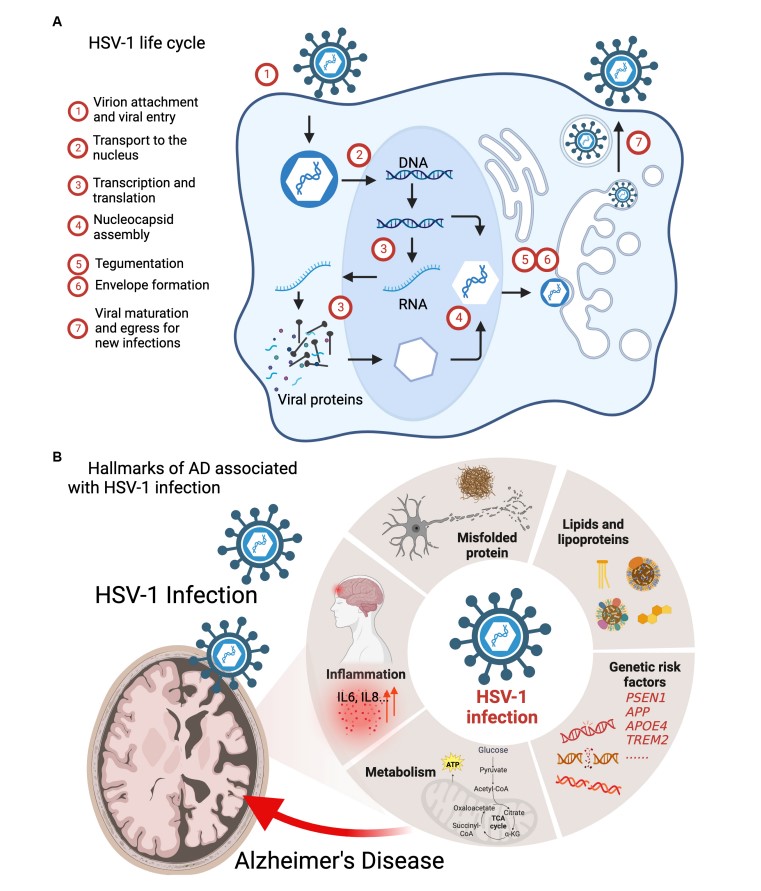 Fig.1 HSV-1 infection interferes the pathogenic processes of AD.1
Fig.1 HSV-1 infection interferes the pathogenic processes of AD.1
Mounting evidence suggests that HSV-1 exacerbates neurodegenerative processes through several pathways.
HSV-1 as a Catalyst for Amyloid Beta (Aβ) Aggregation
HSV-1 infection directly accelerates Aβ accumulation, a defining feature of AD. The viral glycoprotein gB binds heparan sulfate proteoglycans (HSPGs) on neuronal membranes, enhancing cleavage of amyloid precursor protein (APP) by β-secretase (BACE1) to generate neurotoxic Aβ fragments. Concurrently, HSV-1 upregulates presenilin-1 (PSEN1), a key component of the γ-secretase complex, further skewing APP processing toward Aβ production. Viral DNA has been detected within Aβ plaques in postmortem AD brains, suggesting HSV-1 acts as a "seed" for amyloid aggregation. Notably, APOE4 carriers—a high-risk AD cohort—exhibit amplified HSV-1 replication in the brain due to impaired antiviral responses, creating a feedforward loop of Aβ deposition and neuronal damage.
HSV-1's Hidden Role in Neurofibrillary Tangle Formation
Beyond Aβ, HSV-1 exacerbates tau pathology, a driver of cognitive decline in AD. The virus activates glycogen synthase kinase-3β (GSK-3β) via ER stress signaling, phosphorylating tau at critical residues (e.g., Ser202/Thr205), which promotes its dissociation from microtubules and aggregation into neurofibrillary tangles. HSV-1 also inhibits protein phosphatase 2A (PP2A), the primary enzyme responsible for tau dephosphorylation, locking tau in a hyperphosphorylated state. Intriguingly, HSV-1-induced tau pathology occurs independently of Aβ, implicating the virus as a dual-threat instigator of AD's two core pathologies.
HSV-1's Assault on Neuronal Redox Homeostasis
HSV-1 infection unleashes a storm of oxidative damage in the brain. The virus disrupts mitochondrial dynamics, fragmenting networks and impairing electron transport chain complexes, which elevates reactive oxygen species (ROS) production. Viral proteins like ICP0 sequester antioxidant enzymes such as superoxide dismutase 1 (SOD1), leaving neurons vulnerable to ROS-mediated lipid peroxidation and DNA damage. Additionally, HSV-1 depletes glutathione (GSH) and upregulates NADPH oxidase (NOX), further amplifying oxidative stress. These processes converge to activate redox-sensitive kinases (e.g., JNK, p38 MAPK) that drive both Aβ and tau pathologies, creating a self-reinforcing cycle of neurodegeneration.
HSV-1's Subversion of Neuronal Survival Pathways
HSV-1 tilts the balance between neuronal survival and death by dysregulating apoptosis and autophagy. The virus induces apoptosis through caspase-3 activation via mitochondrial cytochrome c release and upregulation of pro-apoptotic proteins (Bax, Bak). Simultaneously, HSV-1 inhibits autophagy—a critical clearance mechanism for toxic protein aggregates—by phosphorylating and inactivating key regulators like Beclin-1 and ATG5. Viral protein US11 further blocks autophagy by binding to protein kinase R (PKR), preventing the degradation of damaged organelles and misfolded proteins. This dual sabotage of apoptosis and autophagy pathways leaves neurons overwhelmed by proteotoxic stress, accelerating AD progression.
HSV-1's regulation of neuroinflammation
HSV-1 ignites a cascade of neuroinflammatory responses that perpetuate AD pathology. The virus activates microglia and astrocytes via Toll-like receptor 2 (TLR2) and NF-κB signaling, triggering the release of pro-inflammatory cytokines such as IL-6, TNF-α, and IL-1β. Critically, HSV-1 primes the NLRP3 inflammasome, driving caspase-1-mediated pyroptosis and amplifying IL-18 secretion, which exacerbates Aβ and tau toxicity. Persistent inflammation also damages the blood-brain barrier, enabling peripheral immune cells to infiltrate the brain and release matrix metalloproteinases (MMPs) that degrade neuronal extracellular matrices. This chronic inflammatory milieu not only accelerates neurodegeneration but also creates a permissive environment for recurrent viral reactivation.
Reference
- Feng, Shu, et al. "Mechanistic insights into the role of herpes simplex virus 1 in Alzheimer's disease." Frontiers in Aging Neuroscience 15 (2023): 1245904. Distributed under Open Access license CC BY 4.0, without modification.