WDR4 Antibodies
Background
WDR4 as members of the family of WD40 repeat proteins, mainly involved in regulation of cell life activities. The protein encoded by this gene mediates protein-protein interactions by forming the β -propeller domain and plays a key role in ribosomal RNA modification and tRNA methylation processes. Its functional abnormalities are directly related to primary microcephaly and brain development defects in humans. In 2015, researchers first analyzed the molecular mechanism involved in regulating tRNA modification through cryo-electron microscopy technology. This highly conserved protein complex has become an important model for studying gene expression regulation and neurodevelopmental diseases, providing a molecular basis for understanding epigenetic regulation and maintaining cellular homeostasis.
Structure of WDR4
WDR4 is a protein with a molecular weight of approximately 36.5 kDa. Its size remains relatively stable among different mammals, but there are subtle differences.
| Species | Human | Mouse | Bovine | Zebrafish |
| Molecular Weight (kDa) | 36.5 | 36.3 | 36.6 | 35.8 |
| Primary Structural Differences | Contains seven WD40 repeating domains | Highly homologous to humans | Core structure domain highly conservative | The number of duplicate domains varies slightly |
The WDR4 protein is composed of approximately 330 amino acids, and its core feature is a typical β -propeller three-dimensional spatial structure formed by multiple WD40 repeat sequences. Each repeating unit contains approximately 40 amino acids, forming reverse parallel β -sheets as "leaves". These "leaves" surround to form a central channel. This domain acts as a molecular scaffold, specifically recognizing and binding to substrate proteins, such as methyltransferase complexes, thereby playing a key regulatory role in tRNA modification.
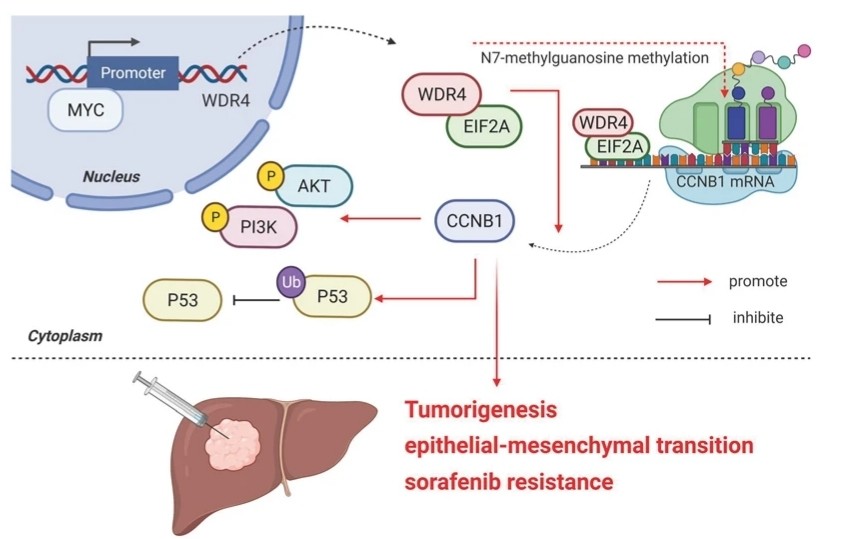 Fig. 1 Model of the regulatory mechanisms of WDR4 in HCC.1
Fig. 1 Model of the regulatory mechanisms of WDR4 in HCC.1
Key structural properties of WDR4:
- Typical β-propeller conformation in 3D space
- Seven WD40 repeat units constitute the protein interaction interface
- Conservative central channel is responsible for substrate recognition and combination
Functions of WDR4
The core function of the WDR4 gene is to participate in tRNA modification and cell cycle regulation. However, it also involves multiple cellular processes, including the maintenance of genomic stability and the regulation of embryonic development.
| Function | Description |
| Regulation of tRNA modification | As a complex scaffold mediates the N2-dimethylation of adenosine 26 of tRNA, it affects the translation efficiency and accuracy. |
| Cell cycle process | By regulating the stability of mitotic spindle checkpoint proteins, normal cell division can be ensured. |
| Genomic stability | Maintain the tRNA modification level in the telomere region to prevent abnormal DNA recombination and chromosomal aneuploidy. |
| Neurodevelopmental regulation | Through epigenetic mechanisms in the development of cerebral cortex influence the proliferation and differentiation of the neural precursor cells. |
| Tumor suppressive function | The loss of its function can lead to abnormal modifications, activate the translation of proto-oncogenes and promote tumorigenesis. |
Unlike single-function modified enzymes, WDR4 forms a multicomplex by dynamically combining different effector proteins. This structural plasticity enables it to coordinate multiple key biological processes ranging from translation regulation to cell division.
Applications of WDR4 and WDR4 Antibody in Literature
1. Wu, Pei-Rung, et al. "Wdr4 promotes cerebellar development and locomotion through Arhgap17-mediated Rac1 activation." Cell Death & Disease 14.1 (2023): 52. https://doi.org/10.1038/s41419-022-05442-z
The article indicates that WDR4 is an aptamer of the CUL4 E3 ligase complex, and its mutation can lead to cerebellar atrophy. Research has found that WDR4 activates Rac1 by ubiquitinating and degrading Arhgap17, maintaining the proliferation of granular neuron precursors and influencing cerebellar development and motor function. This mechanism provides new ideas for the treatment of related diseases.
2. He, Shaohua, et al. "WDR4 gene polymorphisms increase hepatoblastoma susceptibility in girls." Journal of Cancer 13.12 (2022): 3342. https://doi.org/10.7150/jca.76255
This study is the first to explore the association between the WDR4 gene and the risk of hepatoblastoma in children. Analysis of the Han Chinese population in China found that a single WDR4 gene polymorphism does not significantly affect the overall risk of disease. However, in girls, carrying 2 to 5 risk genotypes simultaneously increases the risk of hepatoblastoma by 2.23 times, indicating that the WDR4 polymorphism combination has a significant gender-specific genetic effect.
3. Burkhalter, Martin D., et al. "Cilia defects upon loss of WDR4 are linked to proteasomal hyperactivity and ubiquitin shortage." Cell Death & Disease 15.9 (2024): 660. https://doi.org/10.1038/s41419-024-07042-5
Studies have shown that the loss of function of the WDR4 gene can disrupt the homeostasis of proteins and ubiquitin, leading to abnormally elevated proteasome activity and subsequently disrupting cilia formation. This mechanism is the cause of brain development defects such as primary microcephaly, and inhibiting proteasome activity can improve the related phenotypes.
4. Deng, Linqing, et al. "WDR4 gene polymorphisms and Wilms tumor susceptibility in Chinese children: A five-center case-control study." Journal of Cancer 14.8 (2023): 1293. https://doi.org/10.7150/jca.83747
This study explored the association between WDR4 gene polymorphism and the risk of nephroblastoma. The results showed that the rs6586250 C>T polymorphism significantly increased the risk of disease, especially in specific subgroups. The rs2156315 CT/TT genotype has a protective effect on some young children. This reveals the potential role of WDR4 gene variations in the occurrence of nephroblastoma.
5. Wang, Guoli, et al. "WDR4 promotes the progression and lymphatic metastasis of bladder cancer via transcriptional down-regulation of ARRB2." Oncogenesis 12.1 (2023): 47. https://doi.org/10.1038/s41389-023-00493-z
Research has found that WDR4 in bladder cancer drives lymph node metastasis and tumor progression by promoting the localization of DDX20 and binding to DDX20 and Egr1, thereby inhibiting the transcription of ARRB2. High expression of WDR4 is associated with poor prognosis and is an independent predictor of lymph node metastasis and a potential therapeutic target.
Creative Biolabs: WDR4 Antibodies for Research
Creative Biolabs specializes in the production of high-quality WDR4 antibodies for research and industrial applications. Our portfolio includes monoclonal antibodies tailored for ELISA, Flow Cytometry, Western blot, immunohistochemistry, and other diagnostic methodologies.
- Custom WDR4 Antibody Development: Tailor-made solutions to meet specific research requirements.
- Bulk Production: Large-scale antibody manufacturing for industry partners.
- Technical Support: Expert consultation for protocol optimization and troubleshooting.
- Aliquoting Services: Conveniently sized aliquots for long-term storage and consistent experimental outcomes.
For more details on our WDR4 antibodies, custom preparations, or technical support, contact us at email.
Reference
- Xia, Peng, et al. "MYC-targeted WDR4 promotes proliferation, metastasis, and sorafenib resistance by inducing CCNB1 translation in hepatocellular carcinoma." Cell death & disease 12.7 (2021): 691. https://doi.org/10.1038/s41419-021-03973-5
Anti-WDR4 antibodies
Products List
 Loading...
Loading...





Hot products 
-
Mouse Anti-ASB9 Recombinant Antibody (1D8) (CBMAB-A0529-LY)

-
Mouse Anti-CRYAB Recombinant Antibody (A4345) (CBMAB-A4345-YC)

-
Mouse Anti-ADIPOR1 Recombinant Antibody (V2-179982) (CBMAB-A1368-YC)

-
Mouse Anti-BZLF1 Recombinant Antibody (BZ.1) (CBMAB-AP705LY)

-
Mouse Anti-AMH Recombinant Antibody (5/6) (CBMAB-A2527-YC)

-
Mouse Anti-ALDOA Recombinant Antibody (A2) (CBMAB-A2316-YC)

-
Mouse Anti-ACTN4 Recombinant Antibody (V2-6075) (CBMAB-0020CQ)

-
Mouse Anti-CDK7 Recombinant Antibody (CBYY-C1783) (CBMAB-C3221-YY)

-
Mouse Anti-ACLY Recombinant Antibody (V2-179314) (CBMAB-A0610-YC)

-
Mouse Anti-ADGRL2 Recombinant Antibody (V2-58519) (CBMAB-L0166-YJ)

-
Mouse Anti-BBS2 Recombinant Antibody (CBYY-0253) (CBMAB-0254-YY)

-
Mouse Anti-CORO1A Recombinant Antibody (4G10) (V2LY-1206-LY806)

-
Mouse Anti-AGK Recombinant Antibody (V2-258056) (CBMAB-M0989-FY)

-
Mouse Anti-BACE1 Recombinant Antibody (CBLNB-121) (CBMAB-1180-CN)

-
Mouse Anti-CASP8 Recombinant Antibody (CBYY-C0987) (CBMAB-C2424-YY)

-
Mouse Anti-AAV-5 Recombinant Antibody (V2-503417) (CBMAB-V208-1369-FY)

-
Mouse Anti-APP Recombinant Antibody (5C2A1) (CBMAB-A3314-YC)

-
Mouse Anti-FLI1 Recombinant Antibody (CBXF-0733) (CBMAB-F0435-CQ)

-
Mouse Anti-GIPC2 Recombinant Antibody (10) (CBMAB-G0476-LY)

-
Rabbit Anti-BAD (Phospho-Ser136) Recombinant Antibody (CAP219) (CBMAB-AP536LY)

- AActivation
- AGAgonist
- APApoptosis
- BBlocking
- BABioassay
- BIBioimaging
- CImmunohistochemistry-Frozen Sections
- CIChromatin Immunoprecipitation
- CTCytotoxicity
- CSCostimulation
- DDepletion
- DBDot Blot
- EELISA
- ECELISA(Cap)
- EDELISA(Det)
- ESELISpot
- EMElectron Microscopy
- FFlow Cytometry
- FNFunction Assay
- GSGel Supershift
- IInhibition
- IAEnzyme Immunoassay
- ICImmunocytochemistry
- IDImmunodiffusion
- IEImmunoelectrophoresis
- IFImmunofluorescence
- IGImmunochromatography
- IHImmunohistochemistry
- IMImmunomicroscopy
- IOImmunoassay
- IPImmunoprecipitation
- ISIntracellular Staining for Flow Cytometry
- LALuminex Assay
- LFLateral Flow Immunoassay
- MMicroarray
- MCMass Cytometry/CyTOF
- MDMeDIP
- MSElectrophoretic Mobility Shift Assay
- NNeutralization
- PImmunohistologyp-Paraffin Sections
- PAPeptide Array
- PEPeptide ELISA
- PLProximity Ligation Assay
- RRadioimmunoassay
- SStimulation
- SESandwich ELISA
- SHIn situ hybridization
- TCTissue Culture
- WBWestern Blot
