ACSL4 Antibodies
Background
ACSL4 is an enzyme protein located in the cytoplasmic membrane, mainly distributed in tissues such as the liver, brain, and reproductive system. This enzyme catalyzes the reaction of combining long-chain fatty acids with coenzyme A, participating in lipid metabolism, membrane phospholipid remodeling, and the biosynthesis process of ceramide-like signaling molecules. The activity regulation of ACSL4 is dependent on cellular energy homeostasis and inflammatory response, especially in the ferroptosis pathway, where this enzyme directly affects the sensitivity of cell membrane lipid peroxidation by promoting the esterification reaction of polyunsaturated fatty acids. Gene mutations of ACSL4 are associated with neurodevelopmental abnormalities such as intellectual disability, epilepsy, and Alpers syndrome. In recent years, it has also been found to play a key role in tumor metabolic reprogramming. The research on the structure and function of ACSL4 continues to reveal the molecular mechanism of lipid metabolism in cell fate determination, providing a new perspective for the treatment of metabolic diseases and tumors.
Structure of ACSL4
ACSL4 (long-chain acyl-CoA synthetase 4) is an enzyme protein with a molecular weight of approximately 78 kDa. This weight may vary slightly depending on different subtypes or post-translational modifications.
| Species | Human | Mouse | Rat |
| Molecular Weight (kDa) | 77.8 | 78.1 | 77.9 |
| Primary Structural Differences | Having multiple variable splicing elements, with a highly conserved structure | Has an extremely high sequence homology with humans | The core catalytic domain is exactly the same |
This protein is composed of approximately 700 amino acids and its primary structure folds into a compact spherical structure containing two core domains (adenosine triphosphate formation domain and fatty acid binding domain). The key feature of the structure of the ACS4 protein is the central ATP binding pocket (Walker A motif), which drives the activation of fatty acids by forming an acyl-adenosine intermediate. The palmitoylation modification site of the enzyme is located at the N-terminus, affecting its membrane localization. Its active site is composed of highly conserved arginine (responsible for binding to the phosphate group of ATP) and histidine (involved in proton transfer during the catalytic process), which jointly coordinate substrate specificity and stabilize the reaction transition state.
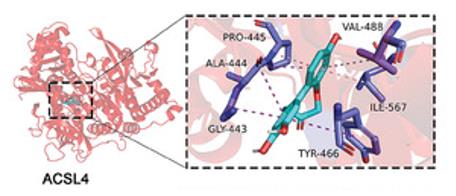 Fig. 1 Molecular docking performed the binding pose details between PrA and ACSL4.1
Fig. 1 Molecular docking performed the binding pose details between PrA and ACSL4.1
Key structural properties of ACSL4:
- Transmembrane helix and membrane-anchoring domain
- Conservative ATP/AMP binding pocket (adenosine acid formation domain)
- Long-chain fatty acid substrate channels and binding sites
- The key amino acids at the active site are involved in the formation and stabilization of the acyl-adenosine intermediate.
Functions of ACSL4
The main function of the protein encoded by the ACSL4 gene is to catalyze the activation of long-chain fatty acids, which is a crucial initial step in lipid metabolism. Additionally, it is involved in various physiological and pathological processes such as membrane phospholipid remodeling, cell signal transduction, and maintenance of energy homeostasis.
| Function | Description |
| Fatty Acid Activation | Catalyzes the combination of long-chain fatty acids with coenzyme A to form fatty acyl coenzyme A, providing a substrate for subsequent β-oxidation or esterification reactions. |
| Membrane Phospholipid Remodeling | By providing specific polyunsaturated fatty acyl coenzyme A substrates, it participates in the renewal of the fatty acid chain composition of cell membrane phospholipids (such as phosphatidylethanolamine). |
| Regulation of Ferroptosis | It is a key positive regulatory factor for ferroptosis (a type of iron-dependent programmed cell death), and its activity and products directly promote lipid peroxidation. |
| Synthesis of Twenty-alkanoic Acid | It provides activated substrates for the metabolism of precursor molecules such as arachidonic acid, influencing the biosynthesis of inflammatory mediators such as prostaglandins and leukotrienes. |
| Energy Metabolism and Storage | By directing fatty acids into the oxidation pathway or the synthesis of triglycerides, it regulates the balance between energy supply within cells and lipid storage. |
Unlike single-functional oxygen carriers, the substrate preference and subcellular localization of ACSL4 determine its guiding role in metabolic flux. Its specific activation of polyunsaturated fatty acids such as arachidonic acid enables it to play a unique "metabolic switch" role in inflammatory and cell death signaling pathways.
Applications of ACSL4 and ACSL4 Antibody in Literature
1. Yang, Yufei, et al. "ACSL3 and ACSL4, distinct roles in ferroptosis and cancers." Cancers 14.23 (2022): 5896. https://doi.org/10.3390/cancers14235896
The article indicates that ACSL3 and ACSL4 are members of the long-chain fatty acid coenzyme A synthase family. They participate in various cancer processes by influencing lipid metabolism and ferroptosis. The regulatory mechanism of these enzymes holds promise for providing new strategies for targeted tumor therapy.
2. Zeng, Kaixuan, et al. "Inhibition of CDK1 overcomes Oxaliplatin resistance by regulating ACSL4‐mediated Ferroptosis in colorectal cancer." Advanced Science 10.25 (2023): 2301088. https://doi.org/10.1002/advs.202301088
The research reveals that CDK1 phosphorylates ACSL4 and promotes its ubiquitination and degradation, thereby inhibiting lipid peroxidation and ferroptosis, and leading to the resistance of colorectal cancer to oxaliplatin. Targeting CDK1 can restore drug sensitivity.
3. Tuo, Qing-zhang, et al. "Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion." Signal transduction and targeted therapy 7.1 (2022): 59. https://doi.org/10.1038/s41392-022-00917-z
This study reveals that thrombin, by activating ACSL4, promotes arachidonic acid metabolism and lipid peroxidation, thereby driving neuronal ferroptosis after ischemic stroke. Targeted inhibition of this pathway can alleviate nerve damage and provides a new strategy for treatment.
4. Cui, Jingxuan, et al. "Protosappanin A protects DOX‐induced myocardial injury and cardiac dysfunction by targeting ACSL4/FTH1 axis‐dependent ferroptosis." Advanced Science 11.34 (2024): 2310227. https://doi.org/10.1002/advs.202310227
This study reveals that PrA derived from hematoxylin directly binds to ACSL4 and FTH1, inhibiting lipid peroxidation and iron ion release, thereby blocking ferroptosis and providing a new target and strategy for the prevention and treatment of anthracycline-induced cardiac toxicity.
5. Feng, Shengjie, et al. "Inhibition of CARM1‐mediated methylation of ACSL4 promotes ferroptosis in colorectal cancer." Advanced Science 10.36 (2023): 2303484. https://doi.org/10.1002/advs.202303484
The research has found that CARM1 methylates the R339 site of ACSL4, promoting its ubiquitination and degradation, thereby inhibiting ferroptosis. Blocking CARM1 can enhance the sensitivity of tumor cells to ferroptosis, providing a new strategy for cancer immunotherapy.
Creative Biolabs: ACSL4 Antibodies for Research
Creative Biolabs specializes in the production of high-quality ACSL4 antibodies for research and industrial applications. Our portfolio includes monoclonal antibodies tailored for ELISA, Flow Cytometry, Western blot, immunohistochemistry, and other diagnostic methodologies.
- Custom ACSL4 Antibody Development: Tailor-made solutions to meet specific research requirements.
- Bulk Production: Large-scale antibody manufacturing for industry partners.
- Technical Support: Expert consultation for protocol optimization and troubleshooting.
- Aliquoting Services: Conveniently sized aliquots for long-term storage and consistent experimental outcomes.
For more details on our ACSL4 antibodies, custom preparations, or technical support, contact us at email.
Reference
- Cui, Jingxuan, et al. "Protosappanin A protects DOX‐induced myocardial injury and cardiac dysfunction by targeting ACSL4/FTH1 axis‐dependent ferroptosis." Advanced Science 11.34 (2024): 2310227. https://doi.org/10.1002/advs.202310227
Anti-ACSL4 antibodies
 Loading...
Loading...





Hot products 
-
Rabbit Anti-AKT3 Recombinant Antibody (V2-12567) (CBMAB-1057-CN)

-
Mouse Anti-CHRNA9 Recombinant Antibody (8E4) (CBMAB-C9161-LY)

-
Mouse Anti-AP4E1 Recombinant Antibody (32) (CBMAB-A2996-YC)

-
Mouse Anti-AMOT Recombinant Antibody (CBYC-A564) (CBMAB-A2552-YC)

-
Mouse Anti-CRYAB Recombinant Antibody (A4345) (CBMAB-A4345-YC)

-
Mouse Anti-BRD3 Recombinant Antibody (CBYY-0801) (CBMAB-0804-YY)

-
Mouse Anti-B2M Recombinant Antibody (CBYY-0050) (CBMAB-0050-YY)

-
Mouse Anti-GGT1 Recombinant Antibody (1F9) (CBMAB-G3273-LY)

-
Mouse Anti-ACO2 Recombinant Antibody (V2-179329) (CBMAB-A0627-YC)

-
Mouse Anti-AQP2 Recombinant Antibody (E-2) (CBMAB-A3358-YC)

-
Rat Anti-C5AR1 Recombinant Antibody (8D6) (CBMAB-C9139-LY)

-
Rat Anti-CD300A Recombinant Antibody (172224) (CBMAB-C0423-LY)

-
Mouse Anti-FLI1 Recombinant Antibody (CBXF-0733) (CBMAB-F0435-CQ)

-
Mouse Anti-BACE1 Recombinant Antibody (61-3E7) (CBMAB-1183-CN)

-
Mouse Anti-CD19 Recombinant Antibody (CBXC-1224) (CBMAB-C1491-CQ)

-
Mouse Anti-ALOX5 Recombinant Antibody (33) (CBMAB-1890CQ)

-
Mouse Anti-CD8 Recombinant Antibody (C1083) (CBMAB-C1083-LY)

-
Mouse Anti-ALDOA Recombinant Antibody (A2) (CBMAB-A2316-YC)

-
Mouse Anti-CAPZB Recombinant Antibody (CBYY-C0944) (CBMAB-C2381-YY)

-
Mouse Anti-BAD (Phospho-Ser136) Recombinant Antibody (CBYY-0138) (CBMAB-0139-YY)

- AActivation
- AGAgonist
- APApoptosis
- BBlocking
- BABioassay
- BIBioimaging
- CImmunohistochemistry-Frozen Sections
- CIChromatin Immunoprecipitation
- CTCytotoxicity
- CSCostimulation
- DDepletion
- DBDot Blot
- EELISA
- ECELISA(Cap)
- EDELISA(Det)
- ESELISpot
- EMElectron Microscopy
- FFlow Cytometry
- FNFunction Assay
- GSGel Supershift
- IInhibition
- IAEnzyme Immunoassay
- ICImmunocytochemistry
- IDImmunodiffusion
- IEImmunoelectrophoresis
- IFImmunofluorescence
- IGImmunochromatography
- IHImmunohistochemistry
- IMImmunomicroscopy
- IOImmunoassay
- IPImmunoprecipitation
- ISIntracellular Staining for Flow Cytometry
- LALuminex Assay
- LFLateral Flow Immunoassay
- MMicroarray
- MCMass Cytometry/CyTOF
- MDMeDIP
- MSElectrophoretic Mobility Shift Assay
- NNeutralization
- PImmunohistologyp-Paraffin Sections
- PAPeptide Array
- PEPeptide ELISA
- PLProximity Ligation Assay
- RRadioimmunoassay
- SStimulation
- SESandwich ELISA
- SHIn situ hybridization
- TCTissue Culture
- WBWestern Blot
